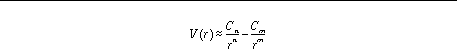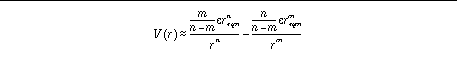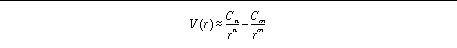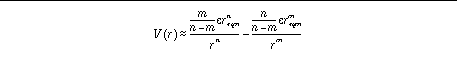III 3. AutoGrid Grid Parameter File: GPF
Extension: .gpf
The input file is often referred to as a "grid parameter file" or "GPF" for short. The scripts described in the appendices give these files the extension ".gpf".
In the grid parameter file, the user must specify the following spatial attributes of the grid maps:
1. the center of the grid map;
2. the number of grid points in each of the
x
-,
y
- and
z
-directions; and
3. the separation or spacing of each grid point.
In addition, the pairwise-atomic interaction energy parameters must be specified. The following lines are required for each ligand atom type,
Y
:
4. the grid map filename for atom type
Y
;
5. seven lines containing the non-bonded parameters for each pairwise-atomic interaction, in the following order:
Y
-C,
Y
-N,
Y
-O,
Y
-S,
Y
-H,
Y
-
X
, (
X
is any other atom type) and
Y
-
M
(
M
is a metal, say).
Using coefficients
C
n
,
C
m
,
n
and
m
, the pairwise interaction energy,
V
(
r
) is given by:
Alternatively, the user can specify reqm, e,
n
and
m
:
This latter method of specification is more intuitive for the user, while
AutoGrid
handles the calculation of the coefficients. By default, the
Y
-
X
and
Y
-
M
lines are copies of the
Y
-H line. But in some systems, such as receptors which consist of DNA/protein complexes, both sulphur
and
phosphorus can be present. In this scenario, the
Y
-
X
line can be used for modeling interactions with receptor-phosphorus atoms. A very rough approximation for phosphorus parameters is to borrow those of carbon.
The "elecmap" line in the grid parameter file is the filename of the electrostatic potential grid map. The following parameter, "dielectric", if negative, indicates that the distance-dependent dielectric function of Mehler and Solmajer
3
will be used. If positive, however, the value of that number will be used as a constant dielectric. For example, if the value were 40.0, then a constant dielectric of 40 would be used.
The
AutoGrid
parameter file format is described below.
AutoGrid Keywords and Commands
receptor <string>
Macromolecule filename, in PDBQ format.
gridfld <string>
The grid field filename, which will be written in a format readable by
AutoDock
and AVS
. The filename extension
must
be `.fld'.
npts <integer> <integer> <integer>
Number of
x
-,
y
- and
z
-grid points. Each
must
be an even integer number. When added to the central grid point, there will be an odd number of points in each dimension. The number of
x
-,
y
- and
z
-grid points need not be equal.
spacing <float>
The grid point spacing, in Å (see the diagram on page 8). Grid points must be uniformly spaced in AutoDock: this value is used in each dimension.
gridcenter <float> <float> <float>
gridcenter auto
The user can explicitly define the center of the grid maps, respectively the
x
,
y
and
z
coordinates of the center of the grid maps (units: Å, Å, Å.) Or the keyword "auto" can be given, in which case
AutoGrid
will center the grid maps on the center of mass of the macromolecule.
types <string>
1-letter names of the atom types present in the ligand;
e.g.
if there are carbons, nitrogens, oxygens and hydrogens, then this line will be "CNOH"; there are no delimiters.
smooth <float>
This is always 0.5Å, when using the AutoDock 3.0 free energy function. It is used to smooth the pairwise atomic affinity potentials (both van der Waals and hydrogen bonds). See the Theory section for more details.
map <string>
Filename of the grid map, for ligand atom type Y; the extension is usually ".map".
nbp_coeffs <float> <float> <integer> <integer>
Either "nbp_coeffs" or "nbp_r_eps" keywords can be used to define Lennard-Jones or hydrogen bond interaction energy parameters. The keyword "nbp_coeffs" specifies coefficients and exponents, in the order
"C
n
C
m
n
m
", delimited by spaces;
n
and
m
are integer exponents. The units of
C
n
and
C
m
must be kcal mol
-1
Å
n
and kcal mol
-1
Å
m
respectively;
n
and
m
have no units.
nbp_r_eps <float> <float> <integer> <integer>
Alternatively, the user can employ "nbp_r_eps" to specify the equilibrium distance and well depth, epsilon, for the atom pair. The equilibrium separation has units of Å and the well depth, epsilon, units of kcal mol
-1
. The integer exponents
n
and
m
must be specified too.
In either case, the order of the parameters must be:
Y
-C,
Y
-N,
Y
-O,
Y
-S,
Y
-H,
Y
-X, and
Y
-M. Repeat 1 "map" line and the 7 "nbp_coeffs"or "nbp_r_eps" lines, for each atom type,
Y
, present in the ligand being docked.
sol_par <float> <float>
This is used to define the atomic fragmental volume and solvation parameters, and should not be changed from the Stouten values used to calibrate the AutoDock 3.0 free energy function.
constant <float>
This is added to all the values in a grid map, and is only set to a non-zero, positive number for hydrogen bonding maps. This value is essentially the penalty for un-formed hydrogen bonds in the complex.
elecmap <string>
Filename for the electrostatic potential energy grid map to be created; filename extension `.map'.
dielectric <float>
Dielectric function flag: if negative,
AutoGrid
will use
distance-dependent
dielectric of Mehler and Solmajer
3
; if the float is positive,
AutoGrid
will use this value as the dielectric constant.
fmap <string>
(Optional.)
Filename for the so-called "floating" grid map
; filename extension `.map'. In such floating grids, the scalar at each grid point is the distance to the nearest atom in the receptor. These values could be used to guide the docking ligand towards the receptor's surface, thus avoiding non-interesting, empty regions.
III 5. Grid Map Field File
Extension: .maps.fld
This is essentially two files in one. It is both an AVS field file, and and
AutoDock
input file with
AutoDock
-specific information `hidden' from AVS in the comments at the head of the file.
AutoDock
uses this file to check that all the maps it reads in are compatible with one-another and itself. For example, in this file, the grid spacing is 0.375 Angstroms, there are 60 intervals in each dimension, the grid is centered near (46,44,14), it was calculated around the macromolecule `
2cpp.pdbqs
', and the
AutoGrid
parameter file used to create this and the maps was `
2cpp.gpf
'. This file also points to a second file, `
2cpp.maps.xyz
', which contains the minimum and maximum extents of the grid box in each dimension,
x, y,
and
z
. Finally, it lists the grid map files that were calculated by
AutoGrid
, here `
2cpp.C.map
', `
2cpp.O.map
' and `
2cpp.e.map
'.
______________________________________________________________________________
# AVS field file
#
# AutoDock Atomic Affinity and Electrostatic Grids
#
# Created by autogrid3.
#
#SPACING 0.375
#NELEMENTS 60 60 60
#CENTER 46.508 44.528 14.647
#MACROMOLECULE 2cpp.pdbqs
#GRID_PARAMETER_FILE 2cpp.gpf
#
ndim=3 # number of dimensions in the field
dim1=61 # number of x-elements
dim2=61 # number of y-elements
dim3=61 # number of z-elements
nspace=3 # number of physical coordinates per point
veclen=3 # number of affinity values at each point
data=float # data type (byte, integer, float, double)
field=uniform # field type (uniform, rectilinear, irregular)
coord 1 file=2cpp.maps.xyz filetype=ascii offset=0
coord 2 file=2cpp.maps.xyz filetype=ascii offset=2
coord 3 file=2cpp.maps.xyz filetype=ascii offset=4
label=C-affinity # component label for variable 1
label=O-affinity # component label for variable 2
label=Electrostatics # component label for variable 2
#
# location of affinity grid files and how to read them
#
variable 1 file=2cpp.C.map filetype=ascii skip=6
variable 2 file=2cpp.O.map filetype=ascii skip=6
variable 3 file=2cpp.e.map filetype=ascii skip=6
______________________________________________________________________________
III 6. AutoDock Docking Parameter File: DPF
Extension: .dpf
AutoDock 3.0
has an interface based on keywords. This is intended to make it easier for the user to set up and control a docking job, and for the programmer to add new commands and functionality. The input file is often referred to as a "docking parameter file" or "DPF" for short. The scripts described in the appendices give these files the extension ".dpf".
All delimiters where needed are white spaces. Default values, where applicable, are given in square brackets [thus]. A comment must be prefixed by the "
#
" symbol, and can be placed at the end of a parameter line, or on a line of its own.
Although ideally it should be possible to give these keywords in any order, not every possible combination has been tested, so it would be wise to stick to the following order.
Command to set the seed for the random number generator
seed <long_integer>
seed time
seed pid
seed <long_integer> <long_integer>
seed time <long_integer>
seed <long_integer> time
seed time pid
seed pid <long_integer>
seed <long_integer> pid
seed pid time
There are two possible random number generator libraries. One is the system's own implementations, and the second is the platform-independent library from the University of Texas Biomedical School. If the user gives just one argument to "
seed
", then AutoDock will use the system's implementation of the random number generator and corresponding system seed call. On most platforms, these are "drand48" and "srand48". The platform-independent library, however, requires two seed values. Giving two arguments to "seed" tells AutoDock to use the platform-independent library for random number generation.
The random-number generator (RNG) for each docking job can be `seeded' with either a user-defined, a time-dependent, or process-ID-dependent seed. These two seeds can be any combination of explicit long integers, the keyword "
time
" or the keyword "
pid
". When two arguments to seed are given, the portable RNG is used; when one is given, the built-in RNG (usually the "drand48" C-function) is used. The portable RNG is required for the genetic algorithm and the Solis and Wets routines. The portable RNG cannot be used with the simulated annealing routine: this needs just one seed parameter. The keyword, "time" gives the number of seconds since the epoch. The epoch is referenced to 00:00:00 CUT (Coordinated Universal Time) 1 Jan 1970. The "pid" gives the UNIX process ID of the currently executing AutoDock process, which is reading this parameter file.
Parameters defining the grid maps to be used
types <string>
Atom names for all atom types present in ligand. Each must be a single character, and only one of: C, N, O, S, H, X, or M. The maximum number of characters allowed in this line is ATOM_MAPS, which is defined in the "autodock.h" include file. Do not use any spaces to delimit the types: they are not needed.
fld <string>
Grid data field file created by
AutoGrid
and readable by
AVS
(must have the extension ".fld").
map <string>
Filename for the first
AutoGrid
affinity grid map of the 1st atom type. This keyword plus filename must be repeated for all atom types in the order specifed by the "
types
" command. In all map files a 6-line header is required, and energies must be ordered according to the nested loops z( y( x ) ).
map <string>
Filename for the electrostatics grid map. 6-line header required, and energies must be ordered according to the nested loops z( y( x ) ).
Parameters defining the ligand and its initial state
move <string>
Filename for the ligand to be docked. This contains most importantly, atom names, xyz-coordinates, anb partial atomic charges in PDBQ format. (Filename extension should be ".pdbq").
about <float> <float> <float>
Use this keyword to specify the center of the ligand,
about
which rotations will be made. (The coordinate frame of reference is that of the ligand PDBQ file.) Usually the rotation center of the ligand is the mean x,y,z-coordinates of the molecule. Inside
AutoDock
, the "
about
" xyz-coordinates are
subtracted
from each atom's coordinates in the input PDBQ file. So internally, the ligand's coordinates become centered at the origin. Units: Å, Å, Å.
tran0 <float> <float> <float>
tran0 random
Initial coordinates for the center of the ligand, in the same frame of reference as the receptor's grid maps. The ligand, which has been internally centered using the "about" coordinates, has the xyz-coordinates of the initial translation "
tran0 x y z
"
added
on. Every run starts the ligand from this location.
Alternatively, the user can just give the keyword "random" and
AutoDock
will pick random initial coordinates instead.
If there are multiple runs defined in this file, using the keyword "runs", then each new run will begin at this same location.
The user
must
specify the absolute
starting
coordinates for the ligand, used to start each run. The user should ensure that the ligand, when translated to these coordinates, still fits within the volume of the grid maps. If there are some atoms which lie outside the grid volume, then AutoDock will automatically correct this, until the ligand is pulled completely within the volume of the grids. (This is necessary in order to obtain complete information about the energy of the initial state of the system.) The user will be notified of any such changes to the initial translation by AutoDock. (Units: Å, Å, Å.)
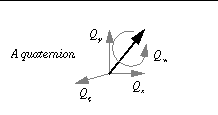
quat0 <float> <float> <float> <float>
quat0 random
[1, 0, 0, 0°]
Respectively: Qx, Qy, Qz
, Q
w.
Initial quaternion (applied to ligand) - Qx, Qy, Qz define the unit vector of the direction of rigid body rotation, and
Q
w
defines the angle of rotation about this unit vector, in ° . (Units: none,none,none, °.)
Alternatively, the user can just give the keyword "random" and
AutoDock
will pick a random unit vector and a random rotation (between 0° and 360°) about this unit vector. Each run will begin at this same random rigid body rotation.
ndihe <integer>
Number of dihedrals or rotatable bonds in the ligand. This may be specifed only if rotatable bonds have been defined using ROOT, BRANCH, TORS
etc
. keywords in the PDBQ
file named on the "
move
" line. The number supplied to this command must agree with the number of torsions defined in this ligand PDBQ file. If this keyword is used, then the next keyword,
dihe0
, must also be specified. Note that if
ndihe
and
dihe0
are not specified and there are defined torsions in the ligand PDBQ file,
AutoDock
assumes that the
chi
1
,
chi
2
,
chi
3
,
etc.
are all zero, and does not change the initial ligand torsion angles. (See also "torsdof" below).
dihe0 <float> ...
Initial
relative
dihedral angles; there must be
ndihe
floating point numbers specified on this line. Each value specified here will be added to the corresponding torsion angle in the input PDBQ file, at the start of each run. Torsion angles are only specified by two atoms, so the definition of rotations is relative. Units: °.
Parameters defining ligand step sizes
tstep <float>
tstep <float> <float>
[2.0 Å]
The first form, with one argument, defines the maximum translation jump for the first cycle that the ligand may make in one simulated annealing step. When "trnrf" is less than 1, the reduction factor is multiplied with the tstep at the end of each cycle, to give the new value for the next cycle. The second form allows the user to specify the value for the first cycle and the last cycle: AutoDock then calculates the reduction factor that satisfies these constraints. Units: Å.
qstep <float>
[50.0°]
Maximum orientation step size for the angular component,
w
, of quaternion. Units: °.
dstep <float>
[50.0°]
Maximum dihedral (torsion) step size. Units: °.
Parameters defining optional ligand torsion constraints
barrier <float>
[10000.0]
(Optional)
This defines the energy-barrier height applied to constrained torsions. When the torsion is at a preferred angle, there is no torsion penalty: this torsion's energy is zero. If the torsion angle falls within a disallowed zone, however, it can contribute up to the full barrier energy. Since the torsion-energy profiles are stored internally as arrays of type `unsigned short', only positive integers between 0 and 65535 are allowed.
gausstorcon <integer> <float> <float>
(Optional)
Adds a constraint to a torsion. The torsion number is identified by an integer. This identifier comes from the list at the top of the AutoTors-generated input ligand PDBQ file (on the REMARK lines). An energy profile will be calculated for this torsion. An inverted Gaussian bell curve is added for each new constraint. To completely specify each Gaussian, two floating point numbers are needed: the
preferred angle
and the
half-width
respectively (both in degrees). Note that the preferred angle should be specified in the range -180
°
to +180
°
; numbers outside this range will be wrapped back into this range. This angle, c, is
relative
to the original torsion angle in the input structure. The
half-width
is the difference between the two angles at which the energy is half the barrier (B/2 in the diagram above). The smaller the half-width, the tighter the constraint.
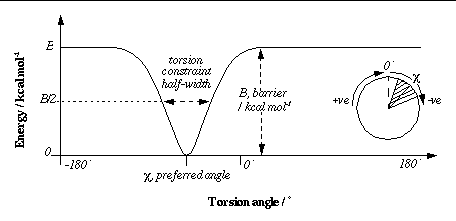
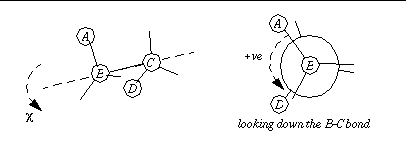
If you wish to constrain to absolute-valued torsion angles, it will be necessary to zero the initial torsion angles in the ligand, before input to
AutoTors
. The problem arises from the ambiguous 2-atom definition of the rotatable bond
B-C
. To identify a torsion angle unambiguously, 4 atoms must be specified:
A-B-C-D
:
The sign convention for torsion angles which we use is anti-clockwise (counter-clockwise) are positive angles, clockwise negative. In the above diagram, looking down the bond
B-C
, the dihedral angle
A-B-C-D
would be positive.
There is no limit to the number of constraints that can be added to a given torsion. Each new torsion-constraint energy profile is combined with the pre-existing one by selecting the minimum energy of either the new or the existing profiles.
showtorpen
(Optional) (Use only with "gausstorcon")
This switches on the storage and subsequent output of torsion energies. During each energy evaluation, the penalty energy for each constrained torsion, as specified by the "gausstorcon" command, will be stored in an array. At the end of each run, the final docked conformation's state variables are output, but with this command, the penalty energy for each torsion will be printed alongside its torsion angle.
hardtorcon <integer> <float> <float>
(Optional)
This command also adds a torsion constraint to the <integer>-th torsion, as numbered in the AutoTors-generated REMARKs. The first float defines the
preferred relative angle
, and the second specifies the
full width
of the allowed range of torsion angles (both in degrees). This type of torsion constraint is "hard" because the torsion is never allowed to take values beyond the range defined. For example, " hardtorcon 3 60. 10." would constrain the third torsion to values between 55° and 65°.
Parameter affecting torsional free energy
torsdof <integer> <float>
[0, 0.3113]
This specifies respectively the number and the coefficient of the torsional degrees of freedom (DOF) for the estimation of the change in free energy upon binding, D
G
binding
. For the purposes of AutoDock 3.0, the number of torsional DOF is the number of rotatable bonds in the ligand, excluding any torsions that rotate one or more hydrogen atoms,
e.g.
hydroxyls, amines, methyls. By default, the coefficient is 0.3113 kcal mol
-1
, although the user can override this as necessary. (Units: none; kcal mol
-1
).
Parameters for ligand internal energies
intnbp_coeffs <float> <float> <integer> <integer>
Respectively: Cn; Cm; n; m. This command specifies the internal pairwise non-bonded energy parameters for flexible ligands, where:
These parameters are needed even if no rotatable bonds were defined in the ligand-PDBQ file. They are only used in the internal energy calculations for the ligand and must be consistent with those used in calculating the grid maps. (Units: kcal mol
-1
Å
n
; kcal mol
-1
Å
m
; none; none, respectively).
intnbp_r_eps <float> <float> <integer> <integer>
Respectively: reqm; e; n; m, This command is an alternative way of specifying the internal pairwise non-bonded energy parameters for flexible ligands, where
AutoDock
calculates the pairwise atomic potential using:
The first two arguments specify the equilibrium distance and well depth, epsilon, for the atom pair. The equilibrium separation has units of Å and the well depth, epsilon, units of kcal mol
-1
. The integer exponents
n
and
m
must be specified too. Obviously,
n
m
. (Units: Å; kcal mol
-1
; none; none, respectively).
intelec
(Optional)
Internal ligand electrostatic energies will be calculated; the products of the partial charges in each non-bonded atom pair are pre-calculated, and output. Note that this is only relevant for flexible ligands.
Parameters for simulated annealing searches
rt0 <float>
[500. cal mol
-1
].
Initial "annealing temperature"; this is actually the absolute temperature multiplied by the gas constant
R
.
R
= 8.314 J mol
-1
K
-1
= 1.987 cal mol
-1
K
-1
. (Units: cal mol
-1
.)
rtrf <float>
Annealing temperature reduction factor,
g
[0.95 cycle
-1
]. See the equation at the bottom of page 5. At the end of each cycle, the annealing temperature is multiplied by this factor, to give that of the next cycle. This must be positive but < 1 in order to cool the system. Gradual cooling is recommended, so as to avoid "
simulated quenching
", which tends to trap systems into local minima.
linear_schedule
schedule_linear
linsched
schedlin
These keywords are all synonymous, and instruct
AutoDock
to use a linear or
arithmetic
temperature reduction schedule during
Monte Carlo
simulated annealing. Unless this keyword is given, a
geometric
reduction schedule is used, according to the
rtrf
parameter just described. If the linear schedule is requested, then any
rtrf
parameters will be ignored. The first simulated annealing cycle is carried out at the annealing temperature
rt0
. At the end of each cycle, the temperature is reduced by (
rt0/cycles
). The advantage of the linear schedule is that the system samples evenly across the temperature axis, which is vital in entropic calculations. Geometric temperature reduction schedules on the other hand, under-sample high temperatures and over-sample low temperatures.
runs <integer>
[10]
Number of automated docking runs.
cycles <integer>
[50]
Number of temperature reduction cycles.
accs <integer>
[100]
Maximum number of accepted steps per cycle.
rejs <integer>
[100]
Maximum number of rejected steps per cycle.
select <character>
[m]
State selection flag. This character can be either
m
for the
minimum
state, or
l
for the
last
state found during each cycle, to begin the following cycle.
trnrf <float>
[1.0]
Per-cycle reduction factor for translations.
quarf <float>
[1.0]
Per-cycle reduction factor for quaternions.
dihrf <float>
Per-cycle reduction factor for dihedrals [1.].
Parameter to set the amount of output
outlev <integer>
[1]
Diagnostic output level. For SA (simulated annealing): 0 = no output, 1 = minimal output, 2 = full state output at end of each cycle; 3 = detailed output for each step. For GA and GA-LS (genetic algorithm-local search): 0 = minimal output, 1 = write minimum, mean, and maximum of each state variable at the end of every generation. Use "outlev 1" for SA, and "outlev 0" for GA and GA-LS. If you use "outlev 1" with GA-LS, you will generate very large log files.
Parameters for trajectory output during SA dockings
trjfrq <integer>
[0]
Output frequency,
n
, for trajectory of ligand, in steps. If
n
= 0, then no trajectory states will be output; otherwise, every
n
th state will be output. The
state
consists of 7 floats describing the
x,y,z
translation, the
x,y,z
components of the quaternion unit vector, the angle of rotation about the quaternion axis; and any remaining floats describing the torsions, in the same order as described in the input ligand PDBQ file).
trjbeg <integer>
[1]
Begin sampling states for trajectory output at the cycle with this value.
trjend <integer>
[50]
End trajectory output at this cycle.
trjout <string>
[lig.trj]
Trajectory filename. AutoDock will write out state variables to this file every "trjfrq" steps. Use the "traj" command in AutoDock's command mode to convert this trajectory of state-variables into a series of PDB frames. The "traj" command is described in § "Using the Command Mode in AutoDock"; see also § "Trajectory Files".
trjsel <string>
[E]
Trajectory output flag, can be either `
A
' or `
E
'; the former outputs only
accepted
steps, while the latter outputs
either
accepted or rejected steps.
watch <string>
(
Optional)
Creates a "watch" file for real-time monitoring of an
in-progress
simulated annealing job. This works only if the "trjfrq" parameter is greater than zero. The watch file will be in PDB format, so give a ".pdb" extension. This file has an exclusive lock placed on it, while AutoDock is writing to it. Once the file is closed, the file is unlocked. This can signal to a watching visualization program that the file is complete and can now be read in, for updating the displayed coordinates. This file is written at exactly the same time as the trajectory file is updated
Parameter for energies of atoms outside the grid
extnrg <float>
[1000.]
External grid energy assigned to any atoms that stray outside the volume of the grid during a docking. Units: kcal mol-1.
Parameter for initializing the ligand in SA
e0max <float> <positive_integer>
[0., 10000]
This is only used by the simulated annealing method. This keyword stipulates that the ligand's initial state cannot have an energy greater than the first value, nor can there be more than the second value's number of retries. Typical energy values range from 0 to 1000 kcal/mol. If the initial energy exceeds this value, a new random state is generated and tested. This process is iterated until the condition is satisfied. This can be particularly useful in preventing runs starting in exceptionally high energy regions. In such cases, the ligand can get trapped because it is unable to take a long enough translational jump. In those grids were the ligand is small enough to fit into the low energy regions with ease, there will not be many iterations before a favorable location is found. But in highly constrained grids, with large ligands, this initialization loop may run almost indefinitely.
Parameters for cluster analysis of docked conformations
rmsref <string>
The root mean square deviation (rmsd) of the docked conformations will be calculated with respect to the coordinates in the PDB or PDBQ file specified here. This is useful when the experimentally determined complex conformation of the ligand is known. The order of the atoms in this file must match that in the input PDBQ file given by the
move
command. These values of rmsd will be output in the last column of the final PDBQ records, after the clustering has been performed.
rmstol <float>
[0.5Å]
rms deviation tolerance for cluster analysis or `structure binning' , carried out after multiple docking runs. If two conformations have an rms less than this tolerance, they will be placed in the same cluster. The structures are ranked by energy, as are the clusters. The lowest energy representative from each cluster is output in PDBQ format to the log file. To keep the ligand's residue number in the input PDBQ file, use the `
-k
' flag; otherwise the clustered conformations are numbered incrementally from 1. (Units: Å).
rmsnosym
When more than one run is carried out in a given job, cluster analysis or `structure binning' will be performed, based on structural rms difference, ranking the resulting families of docked conformations in order of increasing energy. The default method for structure binning allows for atom similarity, as in a tertiary-butyl which can be rotated by +/-120°, but in other cases it may be desirable to bypass this similar atom type checking and calculate the rms on a one-for-one basis. The symmetry checking algorithm scans all atoms in the reference structure, and selects the nearest atom of identical atom type to be added to the sum of squares of distances. This works well when the two conformations are very similar, but this assumption breaks down when the two conformations are translated significantly. Symmetry checking can be turned off using the
rmsnosym
command; omit this command if you still want symmetry checking.
Parameters for re-clustering the results of several jobs
cluster <string>
(Clustering multi-job output only.)
AutoDock
will go into `cluster mode'. Use this command only to perform cluster analysis on the combined output,
<PDBQfilename>
, of several jobs. This command can be very useful when many jobs have been distributed to several machines and run in `parallel'. The docking parameter file will need the following keywords:
rmstol
and
types
; and optionally write_all_cluster_members and/or
rmsnosym
.
It is necessary to
grep
the USER lines along with the ATOM records, since
AutoDock
parses the these lines to determine what the energy of that particular conformation was. For more information, see the example DPF files given later.
write_all_cluster_members
(Clustering multi-job output only.)
This command is used only with the
cluster
command, to write out all members of each cluster instead of just the lowest energy from each cluster. This affects the cluster analysis PDBQ output at the end of each job.
Parameters for genetic algorithm, Lamarckian GA and evolutionary programming searches
ga_pop_size <positive_integer>
[50]
This is the number of individuals in the population. Each individual is a coupling of a genotype and its associated phenotype. Usually, this number is fixed throughout the run. Typical values range from 50 to 200.
ga_num_evals <positive_integer>
[250000]
This is the maximum number of energy evaluations that a GA run should make.
ga_num_generations <positive_integer>
[27000]
This is the maximum number of generations that a GA or LGA run should last.
ga_elitism <integer>
[1]
This is used in the selection mechanism of the GA. This is the number of top individuals that are guaranteed to survive into the next generation.
ga_mutation_rate <float>
[0.02]
This is a floating point number from 0 to 1, representing the probability that a particular gene is mutated. This parameter is typically small.
ga_crossover_rate <float>
[0.80]
This is a floating point number from 0 to 1 denoting the crossover rate. Crossover rate is the expected number of pairs in the population that will exchange genetic material. Setting this value to 0 turns the GA into the evolutionary programming (EP) method, but EP would probably require a concomitant increase in the ga_mutation_rate in order to be effective.
ga_window_size <positive_integer>
[10]
This is the number of preceding generations to take into consideration when deciding the threshold for the worst individual in the current population.
ga_cauchy_alpha <float>
[0]
ga_cauchy_beta <float>
[1]
These are floating point parameters used in the mutation of real number genes. They correspond to the alpha and beta parameters in a Cauchy distribution. Alpha roughly corresponds to the mean, and beta to something like the variance of the distribution. It should be noted, though, that the Cauchy distribution doesn't have finite variance. For the mutation of a real valued gene, a Cauchy deviate is generated and then added to the original value.
Command to set genetic algorithm parameters
set_ga
This command sets the global optimizer to be a genetic algorithm [GA]. This is required to perform a GA search. This passes any '
ga_
' parameters specified
before
this line to the global optimizer object. If this command is omitted, or it is given before the '
ga_
' parameters, your choices will not take effect, and the default values for the optimizer will be used.
To use the traditional genetic algorithm, do not specify the local search parameters, and do not use the "
set_sw1
" or "
set_psw1
" commands.
To use the
Lamarckian genetic algorithm
, you
must
also specify the parameters for local search, and then issue either the '
set_sw1
' or '
set_psw1
' command. The former command uses the strict Solis and Wets local search algorithm, while the latter uses the pseudo-Solis and Wets algorithm: see earlier for details about how they differ.
Parameters for local search
sw_max_its <positive_integer>
[50]
This is the maximum number of iterations that the local search procedure apply to the phenotype of any given individual. This is an unsigned integer. In Bill's experiments, he used a combination of iterations and function evaluations. It seems to me, that a value around 30 should be fine.
sw_max_succ <positive_integer>
[4]
This is the number of successes in a row before a change is made to the rho parameter in Solis & Wets algorithms. This is an unsigned integer and is typically around four.
sw_max_fail <positive_integer>
[4]
This is the number of failures in a row before Solis & Wets algorithms adjust rho. This is an unsigned integer and is usually around four.
sw_rho <float>
[1.0]
This is a parameter of the Solis & Wets algorithms. It defines the initial variance, and specifies the size of the local space to sample.
sw_lb_rho <float>
[0.01]
This is the lower bound on rho, the variance for making changes to genes (
i.e.
translations, orientation and torsions). rho can never be modified to a value smaller than "
sw_lb_rho
".
ls_search_freq <float>
[0.06]
This is the probability of any particular phenotype being subjected to local search.
Commands to choose and set the local search method
Both of these commands, '
set_sw1
' and '
set_psw1
', pass any '
sw_
' parameters set before this line to the local searcher. If you forget to use this command, or give it before the '
sw_
' keywords, your choices will not take effect, and the default values for the optimizer will be used.
set_sw1
Instructs AutoDock to use the classical Solis and Wets local searcher, using the method of uniform variances for changes in translations, orientations and torsions.
set_psw1
Instructs AutoDock to use the pseudo-Solis and Wets local searcher. This method maintains the relative proportions of variances for the translations in Å and the rotations in radians. These are typically 0.2 Å and 0.087 radians to start with, so the variance for translations will always be about 2.3 times larger than that for the rotations (
i.e.
orientation and torsions).
Commands to perform automated docking
simanneal
This command instructs AutoDock to do the specifed number of docking runs using the simulated annealing (SA) search engine. This uses the value set by the "
runs
" keyword as the number of SA docking runs to carry out. All relevant parameters for the simulated annealing job must be set first. These are indicated above by
[SA]
in each keyword description.
do_local_only <integer>
This keyword instructs AutoDock to carry out only the local search of a global-local search; the genetic algorithm parameters are ignored, with the exception of the population size. This is an ideal way of carrying out a minimization using the same force field as is used during the dockings. The "
ga_run
" keyword should not be given. The number after the keyword determines how many dockings will be performed.
do_global_only <integer>
This keyword instructs AutoDock to carry out dockings using only a global search, i.e. the traditional genetic algorithm. The local search parameters are ignored. The "
ga_run
" keyword should not be given. The number after the keyword determines how many dockings will be performed.
ga_run <integer>
[10]
This command invokes the new hybrid, Lamarckian genetic algorithm search engine, and performs the requested number of dockings. All appropriate parameters must be set first: these are listed above by "
ga_
".
Command to perform clustering of docked conformations
analysis
This performs a cluster analysis on results of a docking, and outputs the results to the log file. The docked conformations are sorted in order of increasing energy, then compared by root mean square deviation. If the conformer is within the "rmstol" threshold, it is placed into the same cluster. A histogram is printed showing the number in each cluster, and if more than one member, the cluster's mean energy. Furthermore, a table is printed to the docking log file of cluster rmsd and reference rmsd values.